Atemwegsbasalzellen als Schlüsselzellen in der Pathogenese der Lungenfibrose
Prof. Antje Prasse, Dr. Benedikt Jäger und ihr Team liefern mit ihrer in »Nature Communications« publizierten Arbeit »Airway basal cells show a dedifferentiated KRT17highPhenotype and promote fibrosis in idiopathic pulmonary fibrosis« (https://rdcu.be/cWk1T; DOI: 10.1038/s41467-022-33193-0) wichtige Erkenntnisse zur idiopathischen Lungenfibrose (IPF) – einer tödlichen Krankheit mit begrenzten Behandlungsmöglichkeiten.
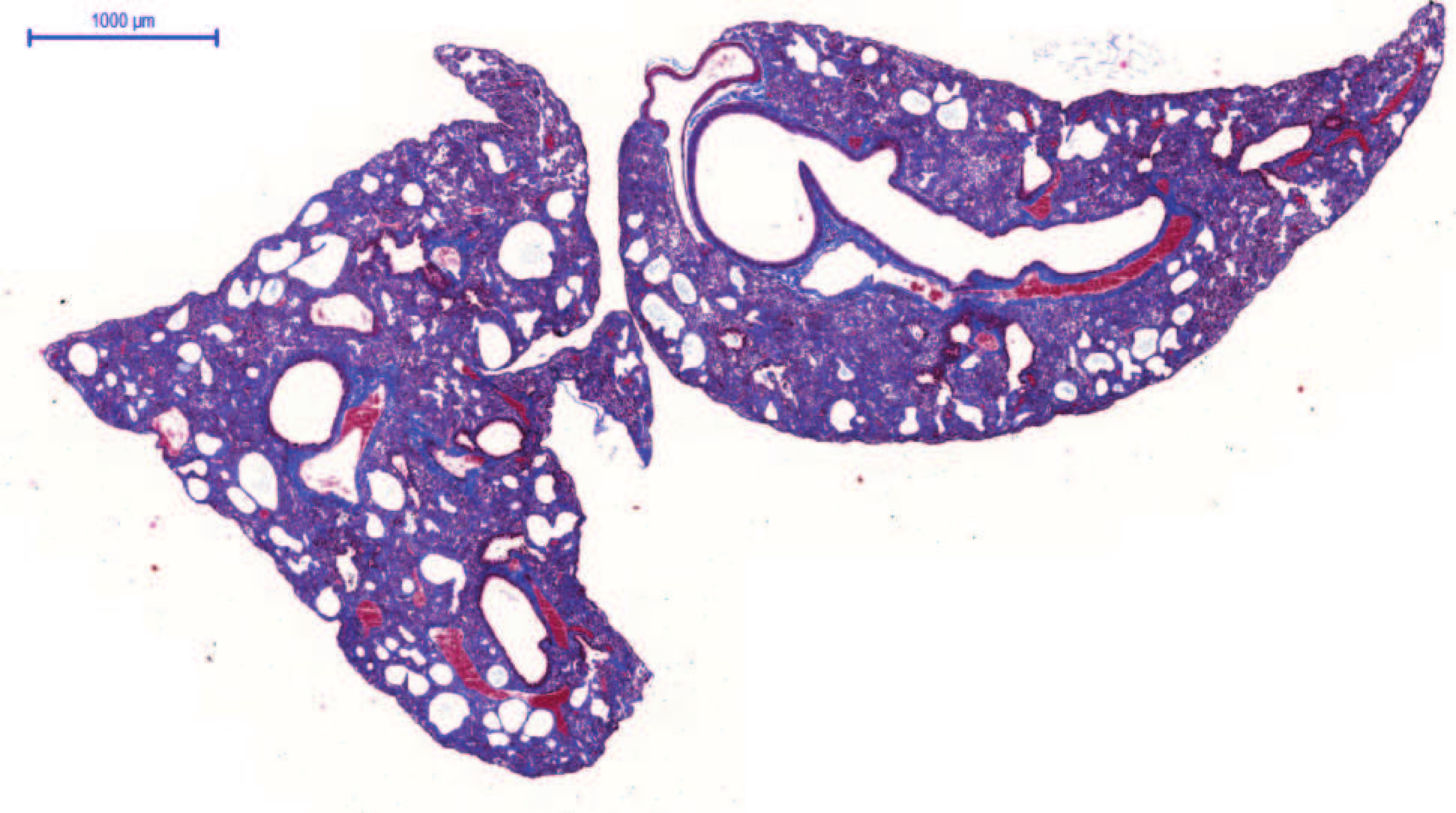
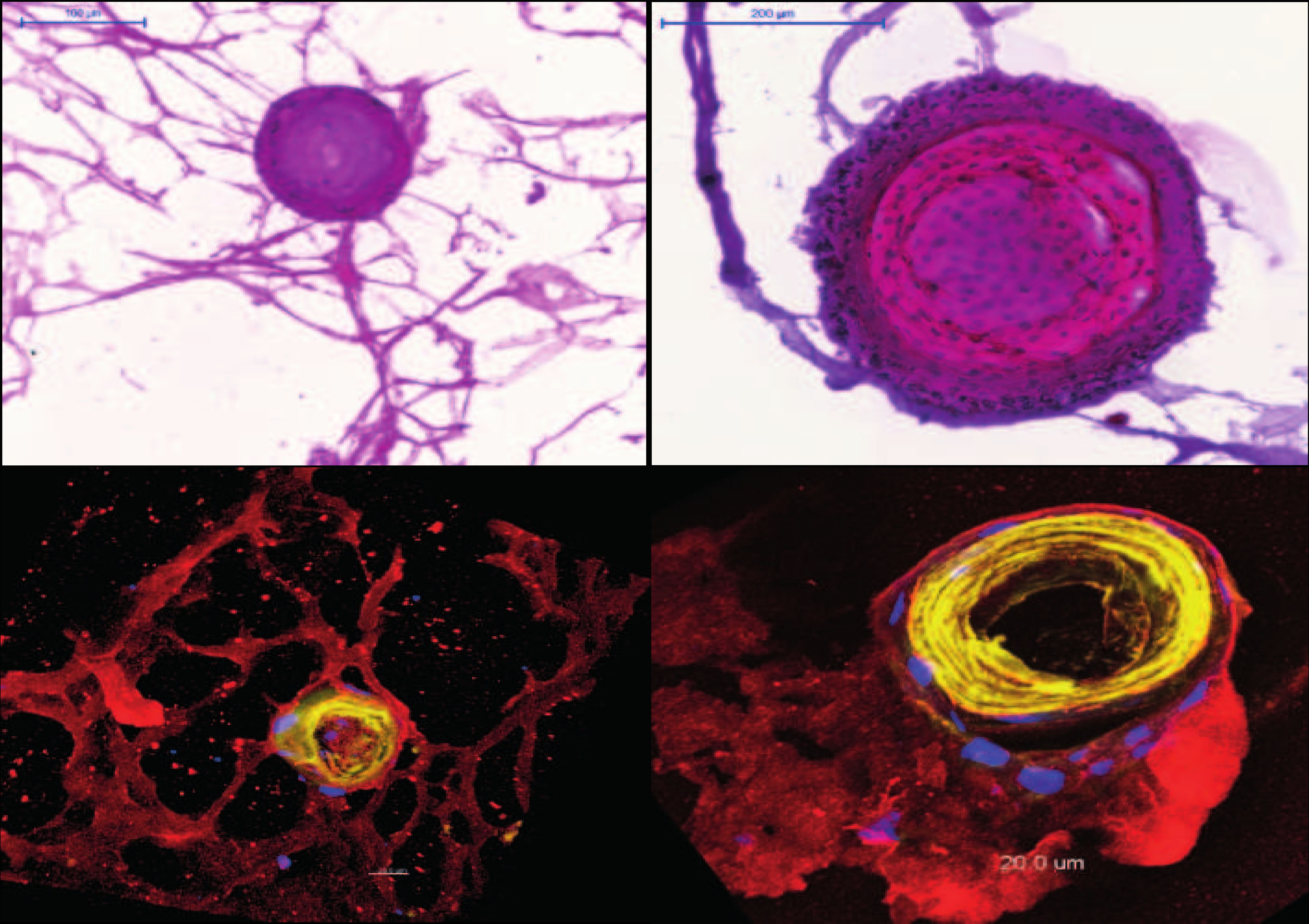
Basalzellen der Atemwege von Patienten mit IPF fördern fibrotische Prozesse aufgrund ihrer atypischen Genexpression und veränderten Differenzierung. Im 3D-Organoid-Modell bilden IPF-Basalzellen im Vergleich zu Basalzellen von gesunden Probanden mehr Organoide sowie De-novo-Bronchialstrukturen, die Lungenentwicklungsprozessen ähneln. Zudem induzieren sie die Proliferation von Fibroblasten und die Ablagerung extrazellulärer Matrix in der Kokultur. Darüber hinaus entwickelte die Arbeitsgruppe ein völlig neuartiges Mausmodell für die IPF, welches auf humanen Basalzellen von IPF-Patienten basiert und damit die Erkrankung viel besser abbildet als bisherige Modelle. Das Forscherteam konnte anhand seiner Transkriptomdatensätze von Patientenzellen mittels bioinformatischer Analysen Prädiktionsmodelle erstellen, die das Therapieansprechen vorhersagen. Aufgrund dieser In-silico-Analysen wurde unter anderem der SRC-Signalweg als ein treibender Faktor identifiziert.
Die Forschenden konnten zeigen, dass der SRC-Inhibitor Saracatinib in ihren neu entwickelten In-vitro- und In-vivo-Modellen die entstehende Fibrose therapeutisch herunterreguliert. Diese Erkenntnisse etablieren Basalzellen als Schlüsselzellen in der Pathogenese der menschlichen idiopathischen Lungenfibrose und damit als neuartiges zelluläres Ziel für die Entwicklung neuer therapeutischer Maßnahmen.